An
elimination reaction is a type of
organic reaction in which two
substituents are removed from a molecule in either a one or two-step mechanism.
[2] The one-step mechanism is known as the
E2 reaction, and the two-step mechanism is known as the
E1 reaction. The numbers do not have to do with the number of steps in the mechanism, but rather the kinetics of the reaction, bimolecular and unimolecular respectively. In rare cases, for molecules possessing particularly poor leaving groups, a third type of reaction,
E1CB, exists.
In most organic elimination reactions, at least one hydrogen is lost to form the double bond: the
unsaturation of the molecule increases. It is also possible that a molecule undergoes
reductive elimination, by which the valence of an atom in the molecule decreases by two, though this is more common in inorganic chemistry. An important class of elimination reactions is those involving
alkyl halides, with good
leaving groups, reacting with a
Lewis base to form an
alkene. Elimination may be considered the reverse of an
addition reaction. When the substrate is asymmetric,
regioselectivity is determined by
Zaitsev's rule or through
Hofmann elimination if the carbon with the most substituted hydrogen is inaccessible.
During the 1920s,
Sir Christopher Ingold proposed a model to explain a peculiar type of chemical reaction: the E2 mechanism. E2 stands for
bimolecular elimination. The reaction involves a one-step mechanism in which
carbon-hydrogen and
carbon-halogen bonds break to form a double bond (
C=C Pi bond).
The specifics of the reaction are as follows:
- E2 is a single step elimination, with a single transition state.
- It is typically undergone by primary substituted alkyl halides, but is possible with some secondary alkyl halides and other compounds.
- The reaction rate is second order, because it's influenced by both the alkyl halide and the base (bimolecular).
- Because the E2 mechanism results in the formation of a pi bond, the two leaving groups (often a hydrogen and a halogen) need to be antiperiplanar. An antiperiplanar transition state has staggeredconformation with lower energy than a synperiplanartransition state which is in eclipsed conformation with higher energy. The reaction mechanism involving staggered conformation is more favorable for E2 reactions (unlike E1 reactions).
- E2 typically uses a strong base. It must be strong enough to remove a weakly acidic hydrogen.
- In order for the pi bond to be created, the hybridizationof carbons needs to be lowered from sp3 to sp2.
- The C-H bond is weakened in the rate determining step and therefore a primary deuterium isotope effectmuch larger than 1 (commonly 2-6) is observed.
- E2 competes with the SN2 reaction mechanism if the base can also act as a nucleophile (true for many common bases).
E1 mechanism
E1 is a model to explain a particular type of chemical elimination reaction. E1 stands for unimolecular elimination and has the following specificities.
- It is a two-step process of elimination: ionization and deprotonation.
- E1 typically takes place with tertiary alkyl halides, but is possible with some secondary alkyl halides.
- The reaction rate is influenced only by the concentration of the alkyl halide because carbocation formation is the slowest step, aka the rate-determining step. Therefore, first-order kinetics apply (unimolecular).
- The reaction usually occurs in the complete absence of a base or the presence of only a weak base (acidic conditions and high temperature).
- E1 reactions are in competition with SN1 reactionsbecause they share a common carbocationic intermediate.
- A secondary deuterium isotope effect of slightly larger than 1 (commonly 1 - 1.5) is observed.
- There is no antiperiplanar requirement. An example is the pyrolysis of a certain sulfonate ester of menthol:
- Only reaction product A results from antiperiplanar elimination. The presence of product B is an indication that an E1 mechanism is occurring.[3]
An example in scheme 2 is the reaction of tert-butylbromide with potassium ethoxide in ethanol.
E1 eliminations happen with highly substituted alkyl halides for two main reasons.
- Highly substituted alkyl halides are bulky, limiting the room for the E2 one-step mechanism; therefore, the two-step E1 mechanism is favored.
- Highly substituted carbocations are more stable than methyl or primary substituted cations. Such stability gives time for the two-step E1 mechanism to occur.
- If SN1 and E1 pathways are competing, the E1 pathway can be favored by increasing the heat.
Specific features : 1 . Rearrangement possible 2 . Independent of concentration and basicity of base
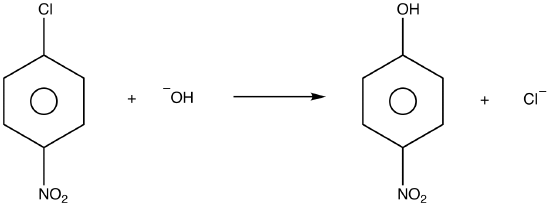
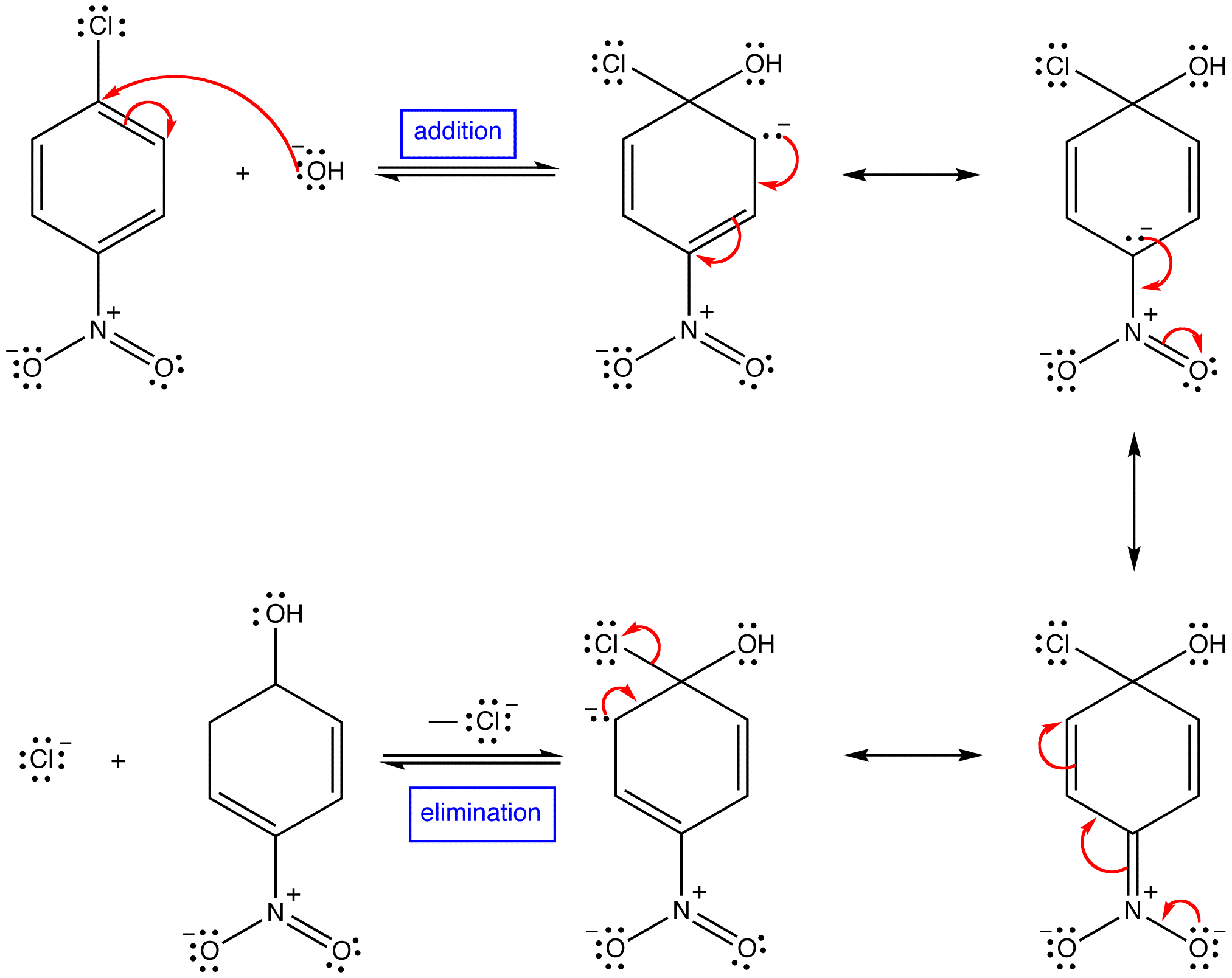
SUBSTITUTION REACTION
Substitution reaction (also known as
single displacement reaction or
single substitution reaction)is a chemical reaction during which one
functional group in a
chemical compound is replaced by another functional group.
[1][2] Substitution reactions are of prime importance in
organic chemistry. Substitution reactions in organic chemistry are classified either as
electrophilicor
nucleophilic depending upon the reagent involved. There are other classifications as well that are mentioned below.
A good example of a substitution reaction is
halogenation. When chlorine gas (Cl-Cl) is irradiated, some of the molecules are split into two chlorine radicals (Cl
.) whose free electrons are strongly
nucleophilic. One of them breaks a weak C-H
covalentbond and grabs the liberated proton to form the electrically neutral H-Cl. The other radical reforms a covalent bond with the CH
3. to form CH
3Cl (
methyl chloride).
|
|
| chlorination of methane by chlorine |
|---|









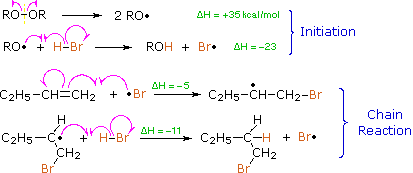
.gif?revision=1&size=bestfit&width=316&height=115)
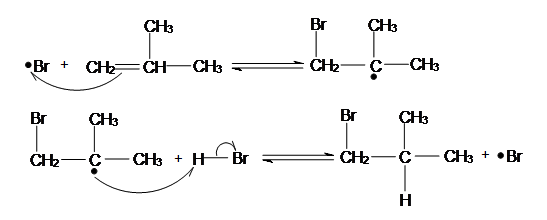 The Bromine Radical will go on and attack the LESS SUBSTITUTED carbon of the alkene. This is because after the bromine radical attacked the alkene a carbon radical will be formed. A carbon radical is more stable when it is at a more substituted carbon due to induction and hyperconjugation. Thus, the radical will be formed at the more substituted carbon, while the bromine is bonded to the less substituted carbon. After a carbon radical is formed, it will go on and attack the hydrogen of a HBr, which a bromine radical will be formed again.
The Bromine Radical will go on and attack the LESS SUBSTITUTED carbon of the alkene. This is because after the bromine radical attacked the alkene a carbon radical will be formed. A carbon radical is more stable when it is at a more substituted carbon due to induction and hyperconjugation. Thus, the radical will be formed at the more substituted carbon, while the bromine is bonded to the less substituted carbon. After a carbon radical is formed, it will go on and attack the hydrogen of a HBr, which a bromine radical will be formed again.





