There is no limit to the number of possible organic reactions and mechanisms.[5][6] However, certain general patterns are observed that can be used to describe many common or useful reactions. Each reaction has a stepwise reaction mechanism that explains how it happens, although this detailed description of steps is not always clear from a list of reactants alone. Organic reactions can be organized into several basic types. Some reactions fit into more than one category. For example, some substitution reactions follow an addition-elimination pathway. This overview isn't intended to include every single organic reaction. Rather, it is intended to cover the basic reactions



Radical reaction
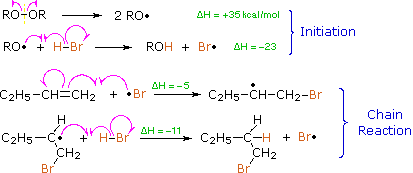
.gif?revision=1&size=bestfit&width=316&height=115)
 The Bromine Radical will go on and attack the LESS SUBSTITUTED carbon of the alkene. This is because after the bromine radical attacked the alkene a carbon radical will be formed. A carbon radical is more stable when it is at a more substituted carbon due to induction and hyperconjugation. Thus, the radical will be formed at the more substituted carbon, while the bromine is bonded to the less substituted carbon. After a carbon radical is formed, it will go on and attack the hydrogen of a HBr, which a bromine radical will be formed again.
The Bromine Radical will go on and attack the LESS SUBSTITUTED carbon of the alkene. This is because after the bromine radical attacked the alkene a carbon radical will be formed. A carbon radical is more stable when it is at a more substituted carbon due to induction and hyperconjugation. Thus, the radical will be formed at the more substituted carbon, while the bromine is bonded to the less substituted carbon. After a carbon radical is formed, it will go on and attack the hydrogen of a HBr, which a bromine radical will be formed again.
Elimination reaction





Addition reaction
An addition reaction, in organic chemistry, is in its simplest terms an organic reaction where two or more molecules combine to form a larger one (the adduct)
Addition reactions are limited to chemical compounds that have multiple bonds, such as molecules with carbon–carbon double bonds (alkenes), or with triple bonds (alkynes). Molecules containing carbon—heterodouble bonds like carbonyl (C=O) groups, or imine (C=N) groups, can undergo addition, as they too have double-bond character.
An addition reaction is the reverse of an elimination reaction. For instance, the hydration of an alkene to an alcohol is reversed by dehydration.
There are two main types of polar addition reactions: electrophilic addition and nucleophilic addition. Two non-polar addition reactions exist as well, called free-radical addition and cycloadditions. Addition reactions are also encountered in polymerizations and called addition polymerization.

General overview of addition reactions. Top to bottom: electrophilic addition to alkene, nucleophilic addition of nucleophile to carbonyl and free-radical addition of halide to alkene
Addition-elimination reaction
In the related addition-elimination reaction an addition reaction is followed by an elimination reaction. In the majority of reactions it involves addition of nucleophiles to carbonyl compounds in what is called nucleophilic acyl substitution.[3]
Other addition-elimination reactions are the reaction of an aliphatic amine to an imine and an aromatic amine to a Schiff base in alkylimino-de-oxo-bisubstitution. The hydrolysis of nitriles to carboxylic acids is also a form of addition-elimination.
1. The overall mechanism of an addition-elimination reaction is known as an addition-elimination mechanism.
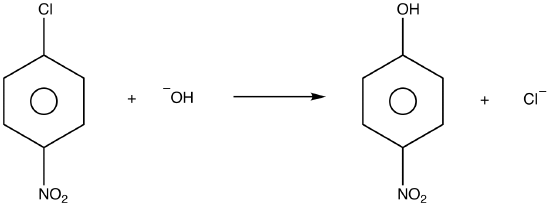
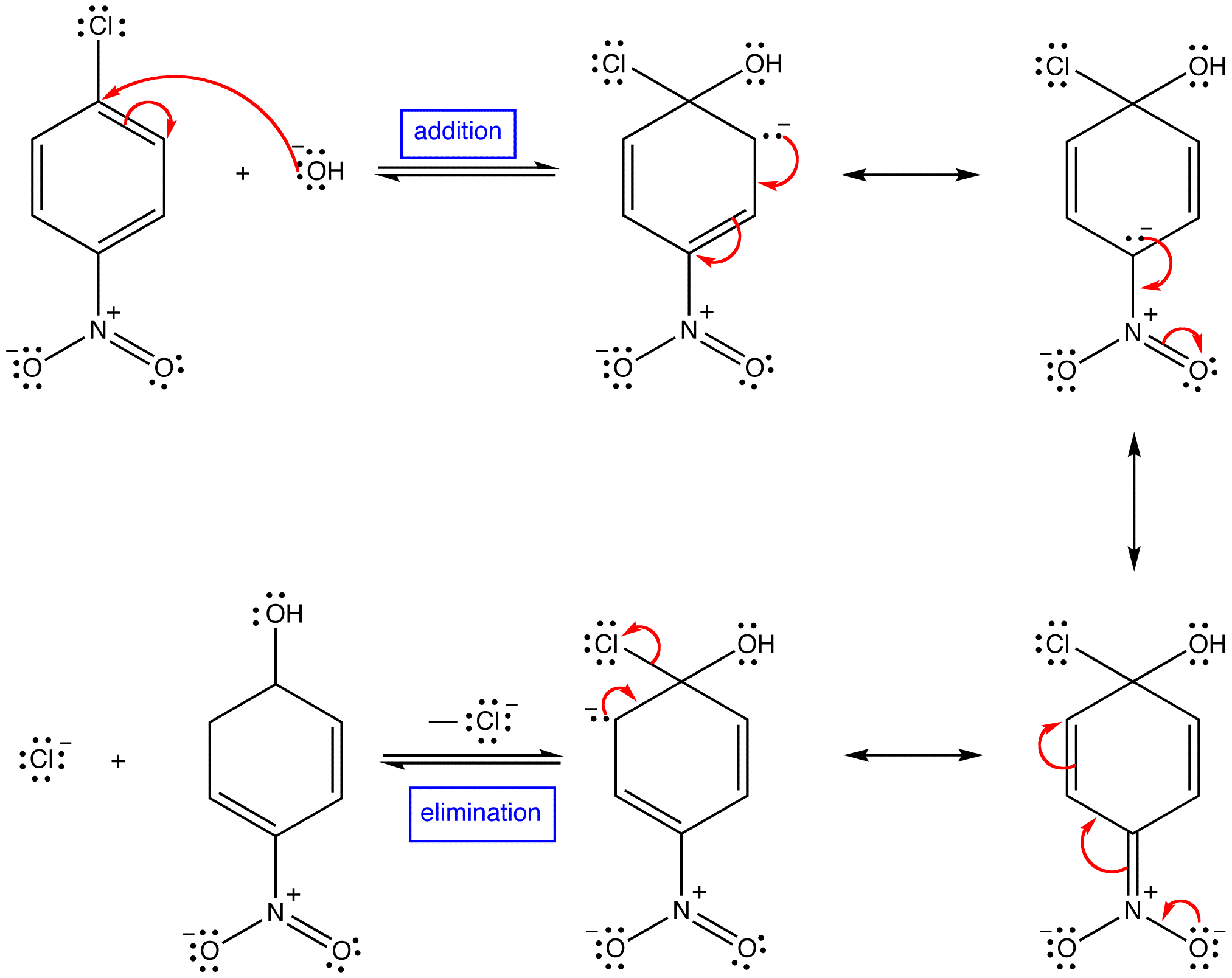
2. Some nucleophilic aromaticsubstitution reactions occur via a two-step mechanism in which the first step, by definition, is an addition and the second step an elimination. The overall mechanism is known as an addition-elimination mechanism.
eg:
mechanism:
Sub types of addition reaction
Electrophilic addition
nucleophilic addition
Radical addition
Electrophilic addition

In organic chemistry, an electrophilic addition reaction is an addition reaction where, in a chemical compound, a π bond is broken and two new σ bonds are formed. The substrate of an electrophilic addition reaction must have a double bond or triple bond.[1]
The driving force for this reaction is the formation of an electrophile X+ that forms a covalent bond with an electron-rich unsaturated C=C bond. The positive charge on X is transferred to the carbon-carbon bond, forming a carbocation during the formation of the C-X bond.

In step 2 of an electrophilic addition, the positively charged intermediate combines with (Y) that is electron-rich and usually an anion to form the second covalent bond.
Step 2 is the same nucleophilic attack process found in an SN1 reaction. The exact nature of the electrophile and the nature of the positively charged intermediate are not always clear and depend on reactants and reaction conditions.
In all asymmetric addition reactions to carbon, regioselectivity is important and often determined by Markovnikov's rule. Organoborane compounds give anti-Markovnikov additions. Electrophilic attack to an aromatic system results in electrophilic aromatic substitution rather than an addition reaction.
Typical electrophilic additions to alkenes with reagents are:
- dihalo addition reactions: X2
- Hydrohalogenations: HX
- Hydration reactions: H2O
- Hydrogenations: H2
- Oxymercuration reactions: mercuric acetate, water
- Hydroboration-oxidation reactions: diborane
- the Prins reaction: formaldehyde, water
Nucleophilic addition
In organic chemistry, a nucleophilic addition reaction is an addition reaction where a chemical compound with an electron-deficient or electrophilic double or triple bond, a π bond, reacts with electron-rich reactant, termed a nucleophile, with disappearance of the double bond and creation of two new single, or σ, bonds. The reactions are involved in the biological synthesis of compounds in the metabolism of every living organism, and are used by chemists in academia and industries such as pharmaceuticals to prepare most new complex organic chemicals, and so are central to organic chemistry. Addition reactions require the presence of groups with multiple bonds in the electrophile: carbon–heteroatom multiple bonds as in carbonyls, imines, and nitriles, or carbon–carbon double or triple bonds.
Addition to carbon–heteroatom double bonds
Nucleophilic addition reactions of nucleophiles with electrophilic double or triple bond (π bonds) create a new carbon center with two additional single, or σ, bonds.[1] Addition of a nucleophile to carbon–heteroatom double or triple bonds such as >C=O or -C
=N show great variety. These types of bonds are polar(have a large difference in electronegativity between the two atoms); consequently, their carbon atoms carries a partial positive charge. This makes the molecule an electrophile, and the carbon atom the electrophilic center; this atom is the primary target for the nucleophile. Chemists have developed a geometric system to describe the approach of the nucleophile to the electrophilic center, using two angles, the Bürgi–Dunitz and the Flippin–Lodge angles after scientists that first studied and described them.[2][3][4]
=N show great variety. These types of bonds are polar(have a large difference in electronegativity between the two atoms); consequently, their carbon atoms carries a partial positive charge. This makes the molecule an electrophile, and the carbon atom the electrophilic center; this atom is the primary target for the nucleophile. Chemists have developed a geometric system to describe the approach of the nucleophile to the electrophilic center, using two angles, the Bürgi–Dunitz and the Flippin–Lodge angles after scientists that first studied and described them.[2][3][4]

This type of reaction is also called a 1,2 nucleophilic addition. The stereochemistry of this type of nucleophilic attack is not an issue, when both alkyl substituents are dissimilar and there are not any other controlling issues such as chelation with a Lewis acid, the reaction product is a racemate. Addition reactions of this type are numerous. When the addition reaction is accompanied by an elimination the reaction type is nucleophilic acyl substitution or an addition-elimination reaction.
Addition to Carbonyl groups
With a carbonyl compound as an electrophile, the nucleophile can be:[1]
- water in hydration to a geminal diol (hydrate)
- an alcohol in acetalisation to an acetal
- a hydride in reduction to an alcohol
- an amine with formaldehyde and a carbonyl compound in the Mannich reaction
- an enolate ion in an aldol reaction or Baylis–Hillman reaction
- an organometallic nucleophile in the Grignard reactionor the related Barbier reaction or a Reformatskii reaction
- ylides such as a Wittig reagent or the Corey–Chaykovsky reagent or α-silyl carbanions in the Peterson olefination
- a phosphonate carbanion in the Horner–Wadsworth–Emmons reaction
- a pyridine zwitterion in the Hammick reaction
- an acetylide in alkynylation reactions.
- a cyanide ion in cyanohydrin reactions
In many nucleophilic reactions, addition to the carbonyl group is very important. In some cases, the C=O double bond is reduced to a C-O single bond when the nucleophile bonds with carbon. For example, in the cyanohydrin reaction a cyanide ion forms a C-C bond by breaking the carbonyl's double bond to form a cyanohydrin.
Addition to Nitriles
- hydrolysis of a nitrile to form an amide or a carboxylic acid
- organozinc nucleophiles in the Blaise reaction
- alcohols in the Pinner reaction.
- the (same) nitrile α-carbon in the Thorpe reaction. The intramolecular version is called the Thorpe–Ziegler reaction.
- Grignard reagents to form imines.[5] The route affords ketones following hydrolysis[6] or primary aminesfollowing imine reduction.[7]
Addition to carbon–carbon double bonds
The driving force for the addition to alkenes is the formation of a nucleophile X− that forms a covalent bond with an electron-poor unsaturated system -C=C- (step 1). The negative charge on X is transferred to the carbon – carbon bond.[1]

In step 2 the negatively charged carbanion combines with (Y) that is electron-poor to form the second covalent bond. Ordinary alkenes are not susceptible to a nucleophilic attack (apolar bond). Styrene reacts in toluene with sodium to 1,3-diphenylpropane [8] through the intermediate carbanion:

Another exception to the rule is found in the Varrentrapp reaction. Fullerenes have unusual double bond reactivity and additions such has the Bingel reaction are more frequent. When X is a carbonyl group like C=O or COOR or a cyanide group (CN), the reaction type is a conjugate addition reaction. The substituent X helps to stabilize the negative charge on the carbon atom by its inductive effect. In addition when Y-Z is an active hydrogen compound the reaction is known as a Michael reaction. Perfluorinated alkenes (alkenes that have all hydrogens replaced by fluorine) are highly prone to nucleophilic addition, for example by fluoride ion from caesium fluoride or silver(I) fluoride to give a perfluoroalkyl anion.
Radical reaction
Protons and other electrophiles are not the only reactive species that initiate addition reactions to carbon-carbon double bonds. Curiously, this first became evident as a result of conflicting reports concerning the regioselectivity of HBr additions. As noted earlier, the acid-induced addition of HBr to 1-butene gave predominantly 2-bromobutane, the Markovnikov Rule product. However, in some early experiments in which peroxide contaminated reactants were used, 1-bromobutane was the chief product. Further study showed that an alternative radical chain-reaction, initiated by peroxides, was responsible for the anti-Markovnikov product. This is shown by the following equations.
The weak O–O bond of a peroxide initiator is broken homolytically by thermal or hight energy. The resulting alkoxy radical then abstracts a hydrogen atom from HBr in a strongly exothermic reaction. Once a bromine atom is formed it adds to the π-bond of the alkene in the first step of a chain reaction. This addition is regioselective, giving the more stable carbon radical as an intermediate. The second step is carbon radical abstraction of another hydrogen from HBr, generating the anti-Markovnikov alkyl bromide and a new bromine atom. Each of the steps in this chain reaction is exothermic, so once started the process continues until radicals are lost to termination events.
This free radical chain addition competes very favorably with the slower ionic addition of HBr described earlier, especially in non-polar solvents. It is important to note, however, that HBr is unique in this respect. The radical addition process is unfavorable for HCl and HI because one of the chain steps becomes endothermic (the second for HCl & the first for HI).
Other radical addition reactions to alkenes have been observed, one example being the peroxide induced addition of carbon tetrachloride shown in the following equation
| RCH=CH2 + CCl4 (peroxide initiator) —> RCHClCH2CCl3 |
The best known and most important use of free radical addition to alkenes is probably polymerization. Since the addition of carbon radicals to double bonds is energetically favorable, concentrated solutions of alkenes are prone to radical-initiated polymerization, as illustrated for propene by the following equation. The blue colored R-group represents an initiating radical species or a growing polymer chain; the propene monomers are colored maroon. The addition always occurs so that the more stable radical intermediate is formed.
RCH2(CH3)CH· + CH3CH=CH2 —>RCH2(CH3)CH-CH2(CH3)CH· + CH3CH=CH2 —>RCH2(CH3)CHCH2(CH3)CH-CH2(CH3)CH· —> etc.
Anti-Markovnikov rule describes the regiochemistry where the substituent is bonded to a less substituted carbon, rather than the more substitued carbon. This process is quite unusual, as carboncations which are commonly formed during alkene, or alkyne reactions tend to favor the more substitued carbon. This is because substituted carbocation allow more hyperconjugation and indution to happen, making the carbocation more stable.
Introduction
This process was first explained by Morris Selig Karasch in his paper: 'The Addition of Hydrogen Bromide to Allyl Bromide' in 1933.1 Examples of Anti-Markovnikov includes Hydroboration-Oxidation and Radical Addition of HBr. A free radical is any chemical substance with unpaired electron. The more substituents the carbon is connected to, the more substituted is that carbon. For example: Tertiary carbon (most substituted), Secondary carbon (medium substituted), primary carbon (least substituted)
Anti-Markovnikov Radical Addition of Haloalkane can ONLY happen to HBr and there MUST be presence of Hydrogen Peroxide (H2O2). Hydrogen Peroxide is essential for this process, as it is the chemical which starts off the chain reaction in the initiation step. HI and HCl cannot be used in radical reactions, because in their radical reaction one of the radical reaction steps: Initiation is Endothermic, as recalled from Chem 118A, this means the reaction is unfavorable. To demonstrate the anti-Markovnikov regiochemistry, I will use 2-Methylprop-1-ene as an example below:
Initiation Steps
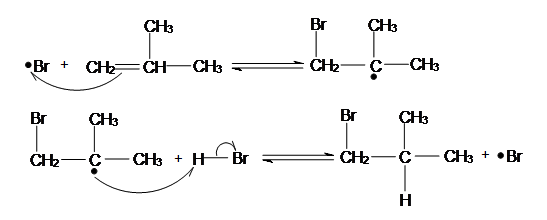
Hydrogen Peroxide is an unstable molecule, if we heat it, or shine it with sunlight, two free radicals of OH will be formed. These OH radicals will go on and attack HBr, which will take the Hydrogen and create a Bromine radical. Hydrogen radical do not form as they tend to be extremely unstable with only one electron, thus bromine radical which is more stable will be readily formed.
Propagation Steps
The Bromine Radical will go on and attack the LESS SUBSTITUTED carbon of the alkene. This is because after the bromine radical attacked the alkene a carbon radical will be formed. A carbon radical is more stable when it is at a more substituted carbon due to induction and hyperconjugation. Thus, the radical will be formed at the more substituted carbon, while the bromine is bonded to the less substituted carbon. After a carbon radical is formed, it will go on and attack the hydrogen of a HBr, which a bromine radical will be formed again.Termination Steps
There are also Termination Steps, but we do not concern about the termination steps as they are just the radicals combining to create waste products. For example two bromine radical combined to give bromine. This radical addition of bromine to alkene by radical addition reaction will go on until all the alkene turns into bromoalkane, and this process will take some time to finish.
References
- K. Peter C. Vollhardt, Neil E. Schore; Organic Chemistry: Structure and Function Fifth Edition; W. H. Freeman and Campany, 2007
- Micheal Vokin; Nuffield Advance Chemistry Student's Book Forth Edition; Person Education Limited, 2004
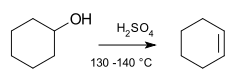
Elimination reaction

An elimination reaction is a type of organic reaction in which two substituents are removed from a molecule in either a one or two-step mechanism.[2] The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers do not have to do with the number of steps in the mechanism, but rather the kinetics of the reaction, bimolecular and unimolecular respectively. In rare cases, for molecules possessing particularly poor leaving groups, a third type of reaction, E1CB, exists.
In most organic elimination reactions, at least one hydrogen is lost to form the double bond: the unsaturation of the molecule increases. It is also possible that a molecule undergoes reductive elimination, by which the valence of an atom in the molecule decreases by two, though this is more common in inorganic chemistry. An important class of elimination reactions is those involving alkyl halides, with good leaving groups, reacting with a Lewis base to form an alkene. Elimination may be considered the reverse of an addition reaction. When the substrate is asymmetric, regioselectivity is determined by Zaitsev's rule or through Hofmann elimination if the carbon with the most substituted hydrogen is inaccessible.
E2 mechanism
During the 1920s, Sir Christopher Ingold proposed a model to explain a peculiar type of chemical reaction: the E2 mechanism. E2 stands for bimolecular elimination. The reaction involves a one-step mechanism in which carbon-hydrogen and carbon-halogen bonds break to form a double bond (C=C Pi bond).
The specifics of the reaction are as follows:
- E2 is a single step elimination, with a single transition state.
- It is typically undergone by primary substituted alkyl halides, but is possible with some secondary alkyl halides and other compounds.
- The reaction rate is second order, because it's influenced by both the alkyl halide and the base (bimolecular).
- Because the E2 mechanism results in the formation of a pi bond, the two leaving groups (often a hydrogen and a halogen) need to be antiperiplanar. An antiperiplanar transition state has staggeredconformation with lower energy than a synperiplanartransition state which is in eclipsed conformation with higher energy. The reaction mechanism involving staggered conformation is more favorable for E2 reactions (unlike E1 reactions).
- E2 typically uses a strong base. It must be strong enough to remove a weakly acidic hydrogen.
- In order for the pi bond to be created, the hybridizationof carbons needs to be lowered from sp3 to sp2.
- The C-H bond is weakened in the rate determining step and therefore a primary deuterium isotope effectmuch larger than 1 (commonly 2-6) is observed.
- E2 competes with the SN2 reaction mechanism if the base can also act as a nucleophile (true for many common bases).

An example of this type of reaction in scheme 1 is the reaction of isobutylbromide with potassium ethoxide in ethanol. The reaction products are isobutylene, ethanoland potassium bromide.
E1 mechanism
E1 is a model to explain a particular type of chemical elimination reaction. E1 stands for unimolecular elimination and has the following specificities.
- It is a two-step process of elimination: ionization and deprotonation.
- Ionization: the carbon-halogen bond breaks to give a carbocation intermediate.
- Deprotonation of the carbocation.
- E1 typically takes place with tertiary alkyl halides, but is possible with some secondary alkyl halides.
- The reaction rate is influenced only by the concentration of the alkyl halide because carbocation formation is the slowest step, aka the rate-determining step. Therefore, first-order kinetics apply (unimolecular).
- The reaction usually occurs in the complete absence of a base or the presence of only a weak base (acidic conditions and high temperature).
- E1 reactions are in competition with SN1 reactionsbecause they share a common carbocationic intermediate.
- A secondary deuterium isotope effect of slightly larger than 1 (commonly 1 - 1.5) is observed.
- There is no antiperiplanar requirement. An example is the pyrolysis of a certain sulfonate ester of menthol:

- Only reaction product A results from antiperiplanar elimination. The presence of product B is an indication that an E1 mechanism is occurring.[3]
- It is accompanied by carbocationic rearrangement reactions

An example in scheme 2 is the reaction of tert-butylbromide with potassium ethoxide in ethanol.
E1 eliminations happen with highly substituted alkyl halides for two main reasons.
- Highly substituted alkyl halides are bulky, limiting the room for the E2 one-step mechanism; therefore, the two-step E1 mechanism is favored.
- Highly substituted carbocations are more stable than methyl or primary substituted cations. Such stability gives time for the two-step E1 mechanism to occur.
- If SN1 and E1 pathways are competing, the E1 pathway can be favored by increasing the heat.
Specific features : 1 . Rearrangement possible 2 . Independent of concentration and basicity of base
SUBSTITUTION REACTION
Substitution reaction (also known as single displacement reaction or single substitution reaction)is a chemical reaction during which one functional group in a chemical compound is replaced by another functional group.[1][2] Substitution reactions are of prime importance in organic chemistry. Substitution reactions in organic chemistry are classified either as electrophilicor nucleophilic depending upon the reagent involved. There are other classifications as well that are mentioned below.
Organic substitution reactions are classified in several main organic reaction types depending on whether the reagent that brings about the substitution is considered an electrophile or a nucleophile, whether a reactive intermediate involved in the reaction is a carbocation, a carbanion or a free radical or whether the substrate is aliphatic or aromatic. Detailed understanding of a reaction type helps to predict the product outcome in a reaction. It also is helpful for optimizing a reaction with regard to variables such as temperature and choice of solvent.
A good example of a substitution reaction is halogenation. When chlorine gas (Cl-Cl) is irradiated, some of the molecules are split into two chlorine radicals (Cl.) whose free electrons are strongly nucleophilic. One of them breaks a weak C-H covalentbond and grabs the liberated proton to form the electrically neutral H-Cl. The other radical reforms a covalent bond with the CH3. to form CH3Cl (methyl chloride).



No comments:
Post a Comment